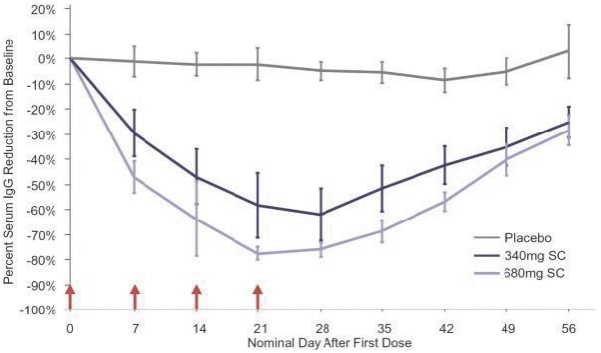
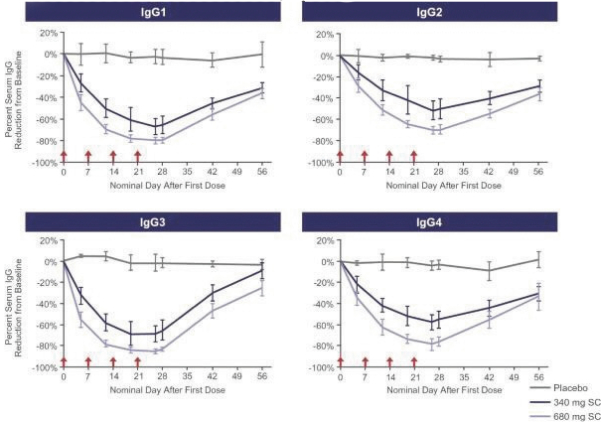
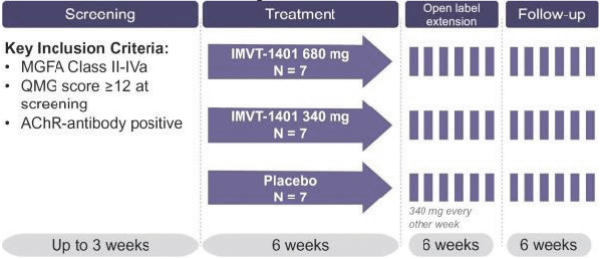
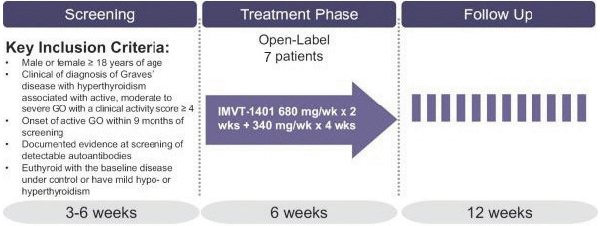
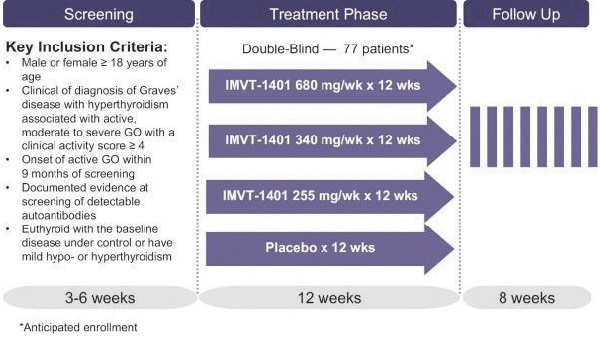
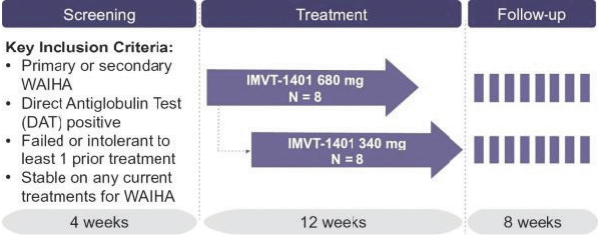
Confidential Treatment Requested by Immunovant, Inc. Pursuant to 17 C.F.R. § 200.83
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell nor does it seek an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED , 2020
PRELIMINARY PROSPECTUS
Shares

Common Stock
We are offering shares of our common stock. Our common stock is listed on the Nasdaq Global Select Market, or Nasdaq, under the symbol “IMVT.” On , 2020, the last reported sale price of our common stock on Nasdaq was $ per share. The final public offering price will be determined through negotiation between us and the lead underwriters in the offering and the recent market price used throughout the prospectus may not be indicative of the actual offering price.
Roivant Sciences Ltd., or RSL or Roivant, is currently our majority stockholder, and we are a “controlled company” within the meaning of the listing rules of Nasdaq. In addition, RSL has the right to elect a certain number of Series A Preferred Directors to our board of directors, in accordance with our amended and restated certificate of incorporation. These directors currently hold a majority of the voting power on all matters presented to the board of directors. See “Description of Capital Stock—Series A Preferred Directors.”
We are an “emerging growth company” as defined under the federal securities laws and, as such, have elected to comply with certain reduced public company reporting requirements for this prospectus and our other filings with the Securities and Exchange Commission.
Investing in our securities involves risks. See “Risk Factors” beginning on page 9.
Neither the Securities and Exchange Commission in the United States nor any other regulatory body has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
| Per Share | Total | |||||||
| Public offering price |
$ | $ | ||||||
| Underwriting discounts and commissions(1) |
$ | $ | ||||||
| Proceeds, before expenses, to us |
$ | $ | ||||||
| (1) | See “Underwriting” for a description of the compensation payable to the underwriters. |
RSL has indicated an interest in purchasing up to $ million of shares of our common stock in this offering on the same terms as those offered to the public. Indications of interest are not binding agreements or commitments to purchase, and the underwriters may determine to sell no shares in this offering to RSL, and RSL may determine to purchase no shares in this offering.
We have granted the underwriters an option for a period of 30 days from the date of this prospectus to purchase up to additional shares of common stock from us, at the public offering price, less the underwriting discounts and commissions. If the underwriters exercise their option in full, the total underwriting discounts and commissions payable by us will be $ , and the total proceeds to us, before estimated expenses, will be $ .
Delivery of the shares of common stock is expected to be made on or about , 2020.
Joint Bookrunning Managers
| SVB Leerink | Guggenheim Securities | Chardan |
Co-Lead Managers
| LifeSci Capital LLC | Baird |
Prospectus dated , 2020